

Humphreys et. al.
It is now relatively trivial to determine the order of amino acids in a protein. Figuring out how that order translates to a complicated three-dimensional structure that performs a specific function, however, is extremely challenging. But after decades of slow progress, Google’s DeepMind AI group announced that it has made tremendous strides toward solving the problem. In July, the system, called AlphaFold, was made open source. At the same time, a group of academic researchers released its own protein-folding software, called RoseTTAFold, built in part using ideas derived from DeepMind’s work.
How effective are these tools? Even if they aren’t as good as some of the statistics suggested, it’s clear they’re far better than anything we’ve ever had. So how will scientists use them?
This week, a large research collaboration set the software loose on a related problem: how these individual three-dimensional structures come together to form the large, multi-protein complexes that perform some of the most important functions in biology.
Beyond 3D
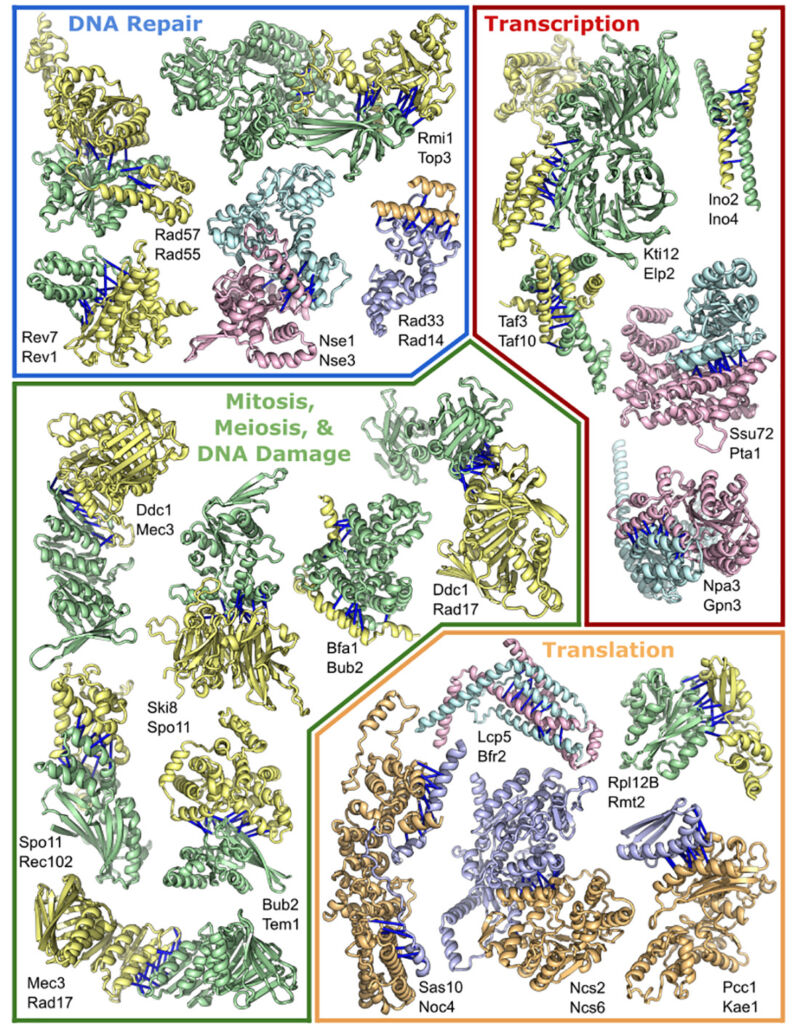
Many individual proteins work just fine on their own, but some aspects of biology require the careful coordination of multiple chemical changes performed as a series of ordered, sequential steps. And for those processes, it’s often easiest for the proteins that need to coordinate to be part of a single complex. For example, the complex that makes copies of our chromosomes typically consists of more than a dozen proteins. Photosystem I, part of plants’ photosynthetic process, is similar in scale. The ribosome, which translates the information in messenger RNAs into the amino acid sequence of proteins, can require over 75 proteins in some species.
Putting these and other complexes together requires the proper folding of their component proteins into the right three-dimensional shapes—the problem that AlphaFold and RoseTTAFold were designed to solve. Once that folding is done, however, the proteins have to interact with each other, fitting together in the right orientation and stabilizing these interactions through contacts among their amino acids (meaning that a positive charge on one protein would be matched by a negative charge on its partner, and so on).
To an extent, the information obtained from AlphaFold and RoseTTAFold should be helpful for this application, because solving the individual structures of proteins should tell us something about the surfaces that could interact. But the methods used by the algorithms turned out to be specifically useful for assembling multi-protein complexes.
RoseTTAFold, for example, solves protein structures in part by chopping their amino acid sequence up into smaller pieces and solving each of them before assembling them into a more complete protein. But the system’s creators found that if RoseTTAFold was given pieces of two different proteins that interact, it would happily assemble both proteins in a way that also captured their interactions, including the right orientation and spacing.
Evolution giveth—and taketh away
The other useful feature is that both algorithms lean heavily on evolution to make their structural predictions. A key step for each is identifying many proteins that are related through common descent and likely to share a common structure. These proteins provide important constraints on the structures that are possible within a given family of related sequences. Certain amino acids interfere with helical structure formation, for example.
Protein complexes can face similar constraints, but there’s an important difference. Let’s say protein A has an amino acid with a positive charge that interacts with a negative one on protein B. If a mutation changes A so that it now has a negative charge, the interaction between the two would be greatly weakened. But protein B could compensate for that issue if a mutation swapped its negative charge for a positive one.
Following pairs of proteins as they change over the course of evolution can provide an indication of whether any changes in one are compensated for by changes in the other. The absence of these sorts of changes can tell us that the proteins are unlikely to interact.
To keep the analysis computationally tractable, the researchers simply paired each protein with everything else in the genome. They found pairwise interactions and later used those interactions to build up larger complexes. However, even knowing potential pairs of interactions left complexes that were limited to a small number of proteins; and attempting to build up something as big as DNA polymerase would have overwhelmed the computational hardware the researchers had access to.






